
BacTermFinder: A Comprehensive and General Bacterial
Terminator Finder using a CNN Ensemble
Seyed Mohammad Amin Taheri Ghahfarokhi
1
and Lourdes Pe˜na-Castillo
∗, 1,2
1
Department of Computer Science, Memorial University of Newfoundland, St. John’s,
Newfoundland, Canada A1B 3X5
2
Department of Biology, Memorial University of Newfoundland, St. John’s, Newfoundland,
Canada A1B 3X9
Abstract
A terminator is a DNA region that ends the transcription process. Currently, multiple computational
tools are available for predicting bacterial terminators. However, these methods are specialized for
certain bacteria or terminator type (i.e., intrinsic or factor-dependent). In this work, we developed
BacTermFinder using an ensemble of Convolutional Neural Networks (CNNs) receiving as input four
different representations of terminator sequences. To develop BacTermFinder, we collected roughly 41k
bacterial terminators (intrinsic and factor-dependent) of 22 species with varying GC-content (from 28% to
71%) from published studies that used RNA-seq technologies. We evaluated BacTermFinder’s performance
on terminators of five bacterial species (not used for training BacTermFinder) and two archaeal species.
BacTermFinder’s performance was compared with that of four other bacterial terminator prediction
tools. Based on our results, BacTermFinder outperforms all other four approaches in terms of average
recall without increasing the number of false positives. Moreover, BacTermFinder identifies both types
of terminators (intrinsic and factor-dependent) and generalizes to archaeal terminators. Additionally, we
visualized the saliency map of the CNNs to gain insights on terminator motif per species. BacTermFinder
is publicly available at https://github.com/BioinformaticsLabAtMUN/BacTermFinder.
1 Background
Transcription starts at the promoter region and ends
at the terminator region. A terminator is a DNA seg-
ment that indicates the end of a gene or operon [55].
The termination of transcription is a process cru-
cial for the accurate synthesis of RNA. In prokary-
otes, termination can occur through either factor-
dependent or factor-independent mechanisms, with
the latter known as intrinsic termination. Intrin-
sic terminators are a DNA region rich in cytosine
(C) and guanine (G) nucleotides followed by a poly-
∗
To whom correspondence should be addressed. Email:
uridine (U) sequence. The RNA created from the
CG-rich region binds with itself, forming a hairpin
structure which causes the RNA polymerase to stall.
The weak base pairing between the adenine (A) nu-
cleotides of the DNA template and the Us of the RNA
transcript let the transcript to detach from the tem-
plate thus, terminating the transcription [57]. While
intrinsic termination requires only cis-acting elements
on nascent RNAs, factor-dependent termination de-
pends on both cis-acting RNA elements and a protein
such as Rho (ρ) which mediates termination via three
distinct mechanisms [62].
Identifying terminators is crucial for understand-
ing operon structure and transcriptional regulation.
1
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint
As both terminator types rely, at least partially,
on cis-acting elements on the DNA template, they
can be identified from genomic sequence. This can
be done via computational identification. Genome-
wide terminator identification via computational ap-
proaches is more affordable and efficient than via wet
lab approaches. Since the development of TransTer-
mHP [36], which is arguably the most cited termi-
nator prediction tool, there have been several com-
putational tools developed for bacterial terminator
prediction (summarized in Table 1). However, as one
can see in Table 1, these tools have either 1) focused
on few (one to three) bacterial species (mostly Es-
cherichia coli and Bacillus subtilis), 2) used a rel-
atively small number of terminators for generating
their model, 3) predicted a single terminator type
(factor-dependent or intrinsic), or 4) a combination of
the above. Thus, there is still the need for a species-
and terminator type-independent tool for predicting
bacterial terminators. The availability of genome-
wide transcription termination sites (TTSs) identi-
fied by RNA-seq technologies such as Term-Seq [14],
Send-seq [33], SMRT-cappable [69], RendSeq [38],
RNATag-seq [60], and dRNA-seq [59] in several bac-
terial species opens the door to generate a species-
agnostic machine learning-based model using a large
number (i.e., thousands) of terminator sequences of
a wide range of bacterial species. Here, we gener-
ated such a method by 1) gathering a large collec-
tion of TTSs from published studies, 2) exploring
thousands of features to represent (encode) termi-
nator sequences, 3) generating and assessing eleven
different machine learning models to identify bac-
terial terminators, and 4) comparatively assessing
the performance of our best model (BacTermFinder)
with the performance of four other bacterial termina-
tor prediction methods (namely, TermNN [8], iTerm-
PseKNC [19], RhoTermPredict [16] and TransTer-
mHP [36]). Our results show that BacTermFinder
can detect intrinsic and factor-dependent terminators
and even archaeal terminators at a higher recall rate
than current tools.
2 Methods
2.1 Terminator sequences collection
We searched the NCBI PubMed [1] and GEO
database [2] using as keywords Term-Seq, Send-seq,
SMRT-cappable, RendSeq, RNATag-seq, and dRNA-
seq to find published studies which identified bac-
terial TTSs. For each identified study, using an
IPython Notebook [37] and pandas data manipula-
tion library [53], we stored in a BED file the genomic
location of identified TTSs (provided in the supple-
mentary material of the corresponding publication).
With BEDTools’ [54] slopeBed and FastaFromBed
commands, we extracted the genomic sequences cor-
responding to 100 nucleotides (nts) flanking the TTSs
(50 nts on either side) into a FASTA file. In each op-
eration, strandedness was taken into account. We
chose 50 nts on either side of the TTSs as a previ-
ous study [44] found intrinsic terminator regions in
E. coli as long as 65 nts. Thus, a sequence of 100 nts
long would be enough to contain this region and al-
low for some variation. Terminators present in plas-
mids were disregarded. We also collected archaeal
TTSs during this process and kept these for the com-
parative assessment. Additionally, we collected TTSs
available in the following databases: RegulonDB [22],
DBTBS [30], and BSGatlasDB [24]. As we collected
multiple data sets for some of the bacterial species,
we needed to remove duplicated sequences from our
data. To achieve this, we merged genomic locations
if they had at least a 60% genomic location overlap.
When the merged sequence was bigger than 100 nts,
we symmetrically removed the extra nts from the be-
ginning and the end of the sequence. Additionally,
sequences containing ambiguous characters (such as
N, Y, etc) were removed. As some of the studies we
collected data from have used different genome as-
semblies for the same bacterium, after getting the ter-
minator sequences, we removed duplicated sequences
(i.e., sequences 100% identical). The bacterial species
with their number of non-redundant terminators used
for training and comparative assessment are listed in
Tables 2 and 3, respectively. Details about each study
are listed in Supplementary Table 1.
2
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint

Table 1: Summary of tools for predicting bacterial terminators. # of terminators indicates how many
terminator sequences were used in the study. The # of species indicates how many bacterial species were
considered. CM is covariance model, DA = discriminant analysis, DL = deep learning, DP = dynamic
programming, and ML = machine learning.
Methods Year Terminator type Method Software available? # of terminators # of species
TermNN [8] 2022 Intrinsic DL Yes 1175 2
ITT pred [26] 2021 Intrinsic Statistical No 137 1
iterb-PPse [18] 2020 Both ML No 928 2
iTerm-PseKNC [19] 2019 Both ML Yes 852 1
RhoTermPredict [16] 2019 Factor-dep. DP Yes 1298 3
OPLS-DA [52] 2018 Factor-dep. DA Yes 104 2
PASIFIC [51] 2017 Intrinsic ML Yes 330 89
RNIE [23] 2011 Intrinsic CM Yes 1062 2
TransTermHP [36] 2007 Intrinsic DP Yes N/A N/A
2.2 Confirming the sequence length
for terminator identification
To confirm that a sequence pattern was within the
100 nts extracted, we used relative nucleotide fre-
quency graphs to visualize whether a distinct pat-
tern was present within this region of interest (ROI).
The relative log nucleotide frequency for a specific
position was calculated by dividing the total count
of each nucleotide in that position by the number of
terminators in that set and then applying a log base
2 function. The log2 ratio of the relative nucleotide
frequency in the ROI vs random genomic regions for
each position and nucleotide was obtained by sub-
tracting from the relative log nucleotide frequency in
the ROI the relative log nucleotide frequency in ran-
domly selected genomic sequences of the same length.
2.3 Non-terminator sequences gener-
ation
To train machine learning-based models, we needed
instances of non-terminators of the same length and
from the same genome as the terminator-containing
sequences. To generate these negative examples, we
used BEDTools’ shuffle command to randomly gener-
ate genomic coordinates different from the terminator
regions. We allowed a maximum sequence overlap
between positive and negative sequences of 20 nts.
A ratio of 1-10 positive to negative was used as it
was shown in [10] that there should be more negative
than positive instances to lower the false-positive rate
during genome scan. Furthermore, the terminator
detection problem has a natural imbalance between
terminator and non-terminator sequences, as the es-
timated number of terminators in a bacterial genome
is relatively tiny compared to the number of all pos-
sible non-terminator sequences of the same length.
Henceforth, we refer to our complete data set (in-
cluding terminator and non-terminator sequences) as
BacTermData.
2.4 Feature engineering and selection
BacTermData contains DNA sequences consisting of
ATCG characters. However, machine learning meth-
ods expect to receive a numerical representation of
the sequences as input. There are many approaches
to numerically representing sequences, such as one-
hot (binary) encoding, k-mer frequencies, etc. As
it is not feasible to determine a priori which repre-
sentation would generate the “best-performing” ma-
chine learning model, one needs to try out several
distinct representations. Here, the best-performing
model refers to a model that maximizes a specific
performance metric, such as the F1-score or area un-
der the precision-recall curve (AUPRC). There are
several libraries or software (e.g., MathFeature [7],
iLearnPlus [9], RepDNA [46]) to generate features
from DNA sequences. Here we decided to use iLearn-
3
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint

Table 2: Number of non-redundant terminators per genome accession used for training. There is a total of
36,542 terminator sequences.
Data source Species Genome Accession Terminators GC (%)
[14, 13] Bacillus subtilis 168 AL009126.3 1800 42.90
[63, 49, 39, 24, 32, 27] Bacillus subtilis 168 NC 000964.3 4872 42.90
[39] Caulobacter crescentus NA1000 CP001340.1 341 66.22
[21] Clostridioides difficile 630 CP010905.2 1646 28.63
[20] Dickeya dadantii 3937 NC 014500.1 1786 55.50
[15] Escherichia coli BW25113 CP009273.1 1095 50.06
[39, 32, 33, 58, 69, 3, 12] Escherichia coli str. K-12 substr. MG1655 NC 000913.3 4139 50.07
[64] Pseudomonas aeruginosa PAO1 NC 002516.2 805 65.61
[50] Staphylococcus aureus JKD6009 LR027876.1 978 32.41
[5] Staphylococcus aureus NCTC 8325 NC 007795.1 566 32.40
[61] Streptococcus pneumoniae D39V CP027540.1 747 39.14
[66] Streptococcus pneumoniae TIGR4 NC 003028.3 1810 39.13
[41, 42] Streptomyces avermitilis MA-4680 BA000030.4 2006 69.72
[28, 42] Streptomyces clavuligerus ATCC27064 CP027858.1 1583 71.64
[42] Streptomyces coelicolor M145 NC 003888.3 1308 71.10
[42, 29] Streptomyces griseus NBRC13350 NC 010572.1 2724 71.21
[43, 42] Streptomyces lividans TK24 CP009124.1 1999 71.22
[42] Streptomyces tsukubaensis NBRC108819 CP020700.1 1283 70.85
[39] Vibrio natriegens ATCC 14048 CP009977.1 905 44.66
[39] Vibrio natriegens ATCC 14048 CP009978.1 257 44.10
[34] Vibrio parahaemolyticus RIMD 2210633 NC 004603.1 1852 44.74
[65] Zymomonas mobilis ZM4 = ATCC 31821 CP023715.1 2040 45.67
Plus to represent the sequences in BacTermData, as
it can generate a wide variety of feature sets. Us-
ing iLearnPlus, we computed 6208 features (belong-
ing to 28 feature sets) per sequence. A feature set
contains features created by one feature generation
method (e.g., one-hot encoding). After generating
the features, we applied feature selection methods to
identify informative features.
To measure the importance of the features, we used
two methods: SHAP [47] and Gini measure of Light
Gradient Boosting Machine (LGBM) [35]. We used
an iterative algorithm to drop features: 1) train an
LGBM model with all remaining features and calcu-
late feature importance values using SHAP and Gini
measure, 2) remove features in the bottom 20% after
sorting the features based on their importance as per
both feature importance methods, 3) repeat step 1
and 2 until the average of the AUPRC obtained dur-
ing 10-fold cross-validation decreases. We observed a
decrease in the AUPRC after reaching 1694 features,
down from 6208 features (Fig. 1). We called these
1694 features (belonging to 22 feature sets) the 22-
sets features. Supplementary Table 2 describes the
1694 selected features.
Generating the 1694 features with ILearnPlus is
computationally intensive (roughly, it processes 30
sequences/second in a High-Performance Computing
Cluster (HPC cluster)). To reduce the computational
requirements, we selected the complete six feature
sets with the most important features. These six fea-
tures sets comprised 60% of the 1694 features (Sup-
plementary Table 2) and called the corresponding fea-
tures the 6-sets features. We also selected individual
feature sets such as ENAC and one-hot (OH) encod-
ing which preserve the spatial location of patterns in
the ROI.
2.5 Machine learning approaches
We utilized convolutional neural networks (CNNs)
and fully-connected neural networks (FCNNs) as
they have been shown to perform well in various do-
4
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint

Table 3: Number of non-redundant terminators per species used for comparative assessment for a total of
4,626 and 3,581 bacterial and archaeal terminators, respectively.
Data source Species Genome Accession Terminators % GC Seq. Tech.
Bacteria
[17] Mycobacterium tuberculosis H37Rv AL123456.3 2202 64.69 Term-Seq
[56] Streptococcus agalactiae NEM316 NC 004368.1 655 35.12 dRNA-Seq
[42] Streptomyces gardneri ATCC 15439 CP059991.1 870 70.73 Term-Seq
[11] Synechocystis PCC 6803 NC 000911.1 553 47.04 Term-Seq
[31] Synechocystis PCC 7338 CP054306.1 346 47.14 Term-Seq
Archaea
[6] Haloferax volcanii DS2 NC 013967 1227 65.69 Term-Seq
[45] Methanococcus maripaludis S2 NC 005791 2354 32.63 Term-Seq
Figure 1: Average precision (an estimate of AUPRC)
as a function of the number of features included in the
training set. Each dot indicates an iteration of the
algorithm used to remove non-informative features.
The shaded area is the standard deviation of 10 cross-
validation folds. The vertical dash line indicates the
point where a drop of AUPRC occurred.
mains [40]. We also used a boosting method because
of boosting methods’ ability to handle tabular data
better than other techniques [25]. Table 4 describes
the ML approaches considered. Table 5 describes the
CNNs architecture.
The architecture of the FCNN is a dense layer con-
taining 400 units and, using the ReLU activation
function, is applied to the categorical input. This
is followed by a dropout layer with a rate of 0.3 to
reduce overfitting. This pattern of a dense layer with
400 units and ReLU activation followed by a dropout
layer with a rate of 0.3 is repeated 6 times more. The
output from these repeated layers is then fed into a
dense layer with 200 units and parametric ReLU ac-
tivation, followed by a dropout layer with a rate of
0.4. This configuration is repeated once more before
the final output layer, which consists of a single unit
with a sigmoid activation function to provide a binary
classification output.
2.6 Model generation and assessment
We used stratified Monte Carlo cross-validation (SM-
CCV) [68] (also called repeated random subsampling)
to select the optimal hyperparameters of the neural
network models. Monte Carlo cross-validation is a
method that randomly determines the training and
validation set in different iterations. The positive-to-
negative data ratio is maintained in each iteration as
it is stratified. We used SMCCV with 100 folds and
10 iterations to find the optimal hyper-parameters
for the models. With 100 folds, 99 % of the data
is for training and 1% for testing on each fold. Neu-
ral networks were implemented in TensorFlow version
2.8.0. We used randomized cross-validation with 50
iterations to find the best hyper-parameters for the
LGBM model (Table 6). We used the Python LGBM
implementation version 3.3.3.
We used SMCCV to assess the models’ perfor-
mance, and calculated average precision, recall, and
5
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint

Table 4: Machine learning approaches considered. Feature sets are described in Supplementary Table 2.
# ML Method Features Description
1 CNN PS2, ENAC, OH, 6-sets features Fusion
1
2 CNN OH, 6-sets features Fusion
3 CNN PS2, ENAC, OH, 6-sets features Append
2
4 CNN PS2 Single
3
5 CNN OH Single
6 CNN ENAC Single
7 CNN NCP Single
8 CNN PS2, ENAC, OH, NCP Ensemble
4
9 FCNN 6-sets features FCNN trained on 6-sets features
10 LGBM 6-sets features A LGBM trained on 6-sets features
11 LGBM 22-sets features A LGBM trained on 22-sets features
Train one CNN for each of the listed feature sets followed by fully-connected layer(s).
Train a CNN receiving as input the concatenated listed feature sets followed by fully-connected layer(s).
Train a CNN receiving as input the listed feature set followed by fully-connected layer(s).
Train independent CNNs followed by fully-connected layer(s) on each listed feature set and the combined
prediction is their average output.
F-score per species. We calculated score thresh-
olds for classification that maximize the F0.5, F1,
and F2 scores. Average precision is calculated as
AP =
∑
n
(R
n
− R
n−1
)P
n
where R
n
and P
n
are the
precision and recall at the n
th
threshold. Recall
is the proportion of actual positive instances cor-
rectly identified by a model (
T P
T P +F N
), while preci-
sion measures the proportion of predicted positive
instances that are actually true positives (
T P
T P +F P
)
where T P is the number of true positives, F N is
the number of false negatives, and F P is the number
of false positives. F-score is calculated as F-score =
(1 + β
2
)
2∗P recision∗Recall
β
2
∗P recision+Recall
where β can be 0.5, 1, or
2 for the corresponding F Score.
3 Results and Discussion
3.1 Clearly visible pattern in ROI in
BacTermData
We visualized the log2 ratio of the relative nucleotide
frequency across the ROI for each study data in-
cluded in BacTermData and confirmed the presence
of a clear pattern around the middle of the ROI in all
the data. Fig. 2 shows the aggregated pattern around
the TTS (middle of the sequence) in the termina-
tor sequences in BacTermData. This indicates that
there is indeed a pattern in BacTermData, which ma-
chine learning approaches could learn to identify ter-
minator sequences. BacTermData consists of 41,168
bacterial terminator sequences (36,542 used for train-
ing) of 25 bacterial strains belonging to four phyla:
Pseudomonadota (formerly Proteobacteria), Bacil-
lota (formerly Firmicutes), Actinomycetota (formerly
Actinobacteria) and Cyanobacteriota. To test the
generalization capabilities of our final model, the
terminator sequences of five bacterial strains (Ta-
ble 3), including the only two belonging to the phy-
lum Cyanobacteriota, were left out of training and
used only for the comparative assessment.
3.2 Selecting a best performing
model: BacTermFinder
We assessed various ML approaches and sequence
representation combinations (Table 4). This assess-
6
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint

Table 5: CNN architecture description.
Layer Type Hyperparameter Value
Conv1D filters 64
kernel size 10
activation PReLU
AveragePooling1D pool size 2
Conv1D filters 64
kernel size 10
activation PReLU
AveragePooling1D pool size 2
BatchNormalization - -
Conv1D filters 64
kernel size 10
activation PReLU
Dropout rate 0.1
Flatten - -
Dense (Dropout) units & (rate) 500 (0.3)
activation PReLU
Dense (Dropout) units & (rate) 600 (0.3)
activation PReLU
Dense (Dropout) units & (rate) 600 (0.3)
activation PReLU
Dense (Dropout) units & (rate) 200 (0.4)
activation PReLU
Dense (Dropout) units & (rate) 200 (0.4)
activation PReLU
Dense units 1
activation Sigmoid
ment involved training and evaluating these combi-
nations on the data shown in Table 2 using Stratified
Monte-Carlo Cross Validation (SMCCV). The SM-
CCV results are presented in Table 7. The LGBM
model trained on the 22-sets features achieved perfor-
mance comparable to that of single CNNs, however
computing the 22-sets features is resource-intensive
and time-consuming. During training the models, we
noticed that the single CNN models were performing
well based on average precision and thus, we decided
to build an ensemble with these four models by sim-
ply averaging their outputs. This approach consist-
ing of an ensemble of the four single CNNs (Fig. 3)
Table 6: Best hyperparameters for LightGBM Model
Hyperparameter Value
boosting type goss
colsample bytree 0.8
learning rate 0.1
max depth 7
min child weight 1
n estimators 3000
num leaves 64
objective cross entropy lambda
random state 42
reg lambda 30
unbalance True
n jobs -1
Figure 2: Log2 ratio of the relative nucleotide fre-
quency of all terminator sequences in BacTermData.
achieved an average precision of 0.7080 ± 0.0248, out-
performing all the other approaches considered (Ta-
ble 7). We selected this as our final model and called
it BacTermFinder. Fig. 4 shows the Precision-Recall
curve (PRC) and Receiver Operating Characteristic
curve (ROC) of BacTermFinder over all SMCCV it-
erations. Clearly, BacTermFinder’s performance is
well above a random classifier’s performance (dashed
lines in Fig. 4).
3.3 Effect of GC content on termina-
tor identification
To look further into the variation in performance
across bacterial species, we visualized the average
precision per bacterium vs their GC content and
coloured it by the phylum to see if there was any pat-
tern (Fig. 5). A linear regression line fitted to this
data indicates a relationship between GC content and
7
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint

Figure 3: Depiction of BacTermFinder architecture.
Table 7: SMCCV average precision ± standard de-
viation. The highest SMCCV average precision is
highlighted in bold. See Table 4 for a description of
the ML approaches.
# Approach Average Precision ± s.d.
1 CNN Fusion 0.5955 ± 0.0232
2 CNN Fusion 0.6553 ± 0.0252
3 CNN Append 0.5949 ± 0.0257
4 CNN Single 0.6738 ± 0.0253
5 CNN Single 0.6775 ± 0.0214
6 CNN Single 0.6544 ± 0.0284
7 CNN Single 0.6730 ± 0.0243
8 CNN Ensemble 0.7080 ± 0.0248
9 FCNN 0.5012 ± 0.0319
10 LGBM 0.5933 ± 0.0269
11 LGBM 0.6476 ± 0.0217
average precision: As the GC content increases, the
performance decreases (Spearman correlation value
of -0.46, p-value 0.031). For a 10% increase in GC
content, the average precision decreases by 0.038.
Thus, BacTermFinder tends to achieve higher av-
erage precision in bacteria with lower GC content.
Bacteria with high GC content tend to have more
factor-dependent terminators [17]. Factor-dependent
terminators do not always have the hairpin structure
of intrinsic terminators [67], and thus, their sequence
motif might be weaker. Additionally, the Rho utiliza-
tion (rut) site is 60-90 nts upstream of the termina-
tor [4], and thus, outside our ROI. These two issues
might explain why BacTermFinder’s performance is
lower on high GC bacteria. BacTermFinder’s per-
Figure 4: PRC in blue and ROC in orange of Bac-
TermFinder on the aggregated results of SMCCV it-
erations. Dashed lines show the performance of a
random classifier.
formance is more consistent (with less variation) for
Bacilota and Actinomycetota than for Pseudomon-
adota (Fig. 5). Further investigation is needed to
understand the reasons for this.
3.4 Assessing BacTermFinder’s pre-
dictions on independent valida-
tion data
We tested BacTermFinder on an independent set of
bacterial and archaeal terminators not included in the
training data (Table 3). For this assessment, we used
the confidence threshold for classification that max-
imizes the F2 score. We visually looked at whether
predicted terminators have a log2 ratio of the rela-
tive nucleotide frequency similar to that of experi-
mentally identified terminators. As it can be seen
from Fig. 6 for S. agalactiae, the log2 ratio of the rel-
ative nucleotide frequency of predicted terminators is
similar to that of experimentally identified termina-
tors. The line smoothness of Fig.6(B) is due to be-
ing a larger number of predicted terminators (12,813)
vs experimentally identified terminators (655). The
saliency map (Fig. 6(C)) shows the contribution of
each nucleotide per position in BacTermFinder’s pre-
dictions. Negative numbers reduce the probability
of predicting a terminator, while positive numbers
8
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint

Figure 5: Average precision vs GC content of Bac-
TermFinder SMCCV results per bacterial strain.
Bacterial strains are coloured based on their phylum.
increase the probability of predicting a terminator.
Figs. 7 and 8 show the same visual analysis for the ar-
chaea M. maripaludis and the cyanobacterium Syne-
chocystis PCC 6803, suggesting that BacTermFinder
predicted terminators have similar sequence patterns
to those present in experimentally determined termi-
nators and that BacTermFinder is also suitable for
finding archaeal terminators.
We noticed some commonalities in the terminator
sequence patterns shown in the saliency maps of H.
volcanii, M. tuberculosis, and S. gardneri (Supple-
mentary Figs. 1-3(C)) which are all organisms with
high GC content in their genome. Similarly, the ter-
minator sequence patterns of S. agalactiae and M.
maripaludis as per their saliency maps (Figs. 6(C)
and 7(C)) show an increasing preference for Gs and
Cs peaking around position 43 followed by a long
poly-T. These two organisms have some of the lowest
GC content in our data. Finally, the two Synechocys-
tis have their own distinct shared pattern (Fig. 8(C)
and Supplementary Fig. 4(C)).
3.5 Comparative assessment for
genome-wide terminator predic-
tion
We compared BacTermFinder’s performance
with that of TermNN [8], iTerm-PseKNC [19],
RhoTermPredict [16] and TransTermHP [36] on
terminators of five bacterial species (Table 3) not
used for generating our model. We decided to include
TermNN, RhoTermPredict and iTerm-PseKNC in
the comparative assessment because they are the
most recently developed tools available for predict-
ing intrinsic, factor-dependent and both types of
terminators, respectively (Table 1). We included
TransTermHP because, as mentioned earlier, is the
most cited tool for bacterial terminator prediction.
All programs were used to obtain genome-wide
terminator predictions. As the experimentally
identified terminators in BacTermData are not
exhaustive; i.e., there might be actual terminators
missing from BacTermData. Predictive performance
was evaluated using recall at ten sequence overlap-
ping thresholds between the predicted terminators
and the actual terminators (Table 8). To do this, we
used BEDtools’ intersect command and calculated
recall at each overlap threshold.
We included in the comparative assessment bacte-
ria with different GC content. Bacteria with high GC
content tend to have a larger proportion of factor-
dependent terminators [17]. For example, M. tu-
berculosis is reported to have a large proportion
(up to 54%) of factor-dependent terminators [17].
As RhoTermPredict was designed to predict factor-
dependent terminators, we hypothesized it would
perform better on bacteria with high GC content
than on bacteria with low GC content. On the other
hand, we hypothesized that termNN and TransTer-
mHP would perform better on bacteria with low GC
content than on bacteria with high GC content, as
these two tools were designed to identify intrinsic
terminators. Our results (Table 8) indeed support
these hypotheses. RhoTermPredict achieved its high-
est recall (roughly 24%) on M. tuberculosis and S.
gardneri which are the two organisms with the high-
est proportion of factor-dependent terminators. Bac-
TermFinder’s recall on these two organisms (i.e., M.
9
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint

A
B
C
Figure 6: Visualization of S. agalactiae experimentally identified terminators (A) vs BacTermFinder genome-
wide predictions (B). A. Log2 ratio of the relative nucleotide frequency of experimentally determined termi-
nators. B. Log2 ratio of the relative nucleotide frequency of genome-wide predicted terminators. C. Saliency
map generated using using [48].
10
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint

A
B
C
Figure 7: Visualization of M. maripaludis S2 experimentally identified terminators (A) vs BacTermFinder
genome-wide predictions (B). A. Log2 ratio of the relative nucleotide frequency of experimentally determined
terminators. B. Log2 ratio of the relative nucleotide frequency of genome-wide predicted terminators. C.
Saliency map generated using [48].
11
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint

A
B
C
Figure 8: Visualization of Synechocystis PCC 6803 experimentally identified terminators (A) vs Bac-
TermFinder genome-wide predictions (B). A. Log2 ratio of the relative nucleotide frequency of experimentally
determined terminators. B. Log2 ratio of the relative nucleotide frequency of genome-wide predicted termi-
nators. C. Saliency map generated using [48].
12
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint

Figure 9: Distribution of the number of predicted
terminators per gene across the five bacterial species
in our independent validation data. The horizontal
line inside each box indicates the median value, and
the bottom and top of each box indicate the 25 and
75 percentile, respectively
tuberculosis and S. gardneri) is comparable to or
better than that of RhoTermPredict. All tools but
RhoTermPredict achieved their highest recall on S.
agalactiae (lowest GC content). Tools’ recall var-
ied between species with an overall recall range of
[0.03, 0.82]. BacTermFinder outperformed ITerm-
PseKNC, TermNN and TransTermHP in terms of
mean recall across various overlap thresholds in all
the species in the validation set, and it achieved the
highest mean recall overall (Table 8). This result sug-
gests that BacTermFinder is able to find both types
of terminators (intrinsic and factor-dependent) and
can generalize to phyla not seen during training (e.g.,
Cyanobacteriota).
As we have an incomplete annotation of all ter-
minators in any given bacterial genome, it is hard to
estimate the genome-wide false positive rate of termi-
nator prediction tools since a prediction might indeed
be correct even though a terminator might not have
yet been determined experimentally in that location.
However, the number of predicted terminators per
gene can provide an estimate of the number of false
positives. Figure 9 shows the distribution of num-
ber of terminators predicted per gene by TermNN,
RhoTermPredict and BacTermFinder across the five
bacteria in our independent validation data. Bac-
TermFinder displays less variation in the number of
predicted terminators per gene, and predicts, on aver-
age, 6.62 ± 1.18 terminators per gene; while TermNN
and RhoTermPredict predict 8.89 ± 3.52 and 4.30 ±
3.20, respectively. RhoTermPredict predicts less ter-
minators per gene; however, its recall is substantially
lower than BacTermFinder’s for four out of five bac-
teria in our independent validation data. The results
provided in Figure 9 and Table 8 indicate that Bac-
TermFinder’s false positive rate is lower than that of
TermNN (the 2nd best tool) while achieving a higher
recall rate.
We used TermNN and BacTermFinder, which are
the two tools with the highest recall as per Table 8,
to find archaeal terminators. BacTermFinder out-
performs TermNN in terms of recall in predicting
archaeal terminators (Table 9). This indicates that
BacTermFinder can generalize to identify archaeal
terminators.
3.5.1 BacTermFinder can predict the loca-
tion of terminators accurately
We assessed how closely our predictions aligned with
actual terminator locations. To do this, we visual-
ized the recall rate as a function of sequence overlap
of the predicted terminators with the actual termi-
nators (Fig. 10). We hypothesized that we would ob-
serve a declining trend as we moved towards stricter
overlap thresholds, which is indeed the case. We com-
pared our overlap vs recall with that of TermNN.
Our results show that 1) on every overlap thresh-
old, BacTermFinder outperforms or is comparable
to TermNN, and 2) BacTermFinder’s recall drops at
stricter overlaps than TermNN’s recall. The latter in-
dicates that BacTermFinder can find the location of
terminators more accurately than TermNN. However,
BacTermFinder’s recall sharply decreases at overlap
thresholds higher than 0.8, which suggests the poten-
tial need for a nucleotide-wise segmentation approach
to achieve accuracy at the nucleotide level.
Additionally, we assessed the accuracy of the prob-
13
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint

Table 8: Average recall over ten overlap thresholds ± standard deviation for four other terminator prediction
tools and BacTermFinder. The corresponding genome accession and GC content (%) is provided below each
bacterium name. The highest recall per row is highlighted in bold.
Bacterium RhoTermPredict ITerm-PseKNC TransTermHP TermNN BacTermFinder
Streptomyces gardneri ATCC 15439 0.2499 ± 0.1263 0.0101 ± 0.0042 0.1709 ± 0.1035 0.3690 ± 0.2227 0.6029 ± 0.2177
CP059991.1, GC=70.73%
Mycobacterium tuberculosis H37Rv 0.2424 ± 0.1640 0.1834 ± 0.1149 0.0033 ± 0.0018 0.1396 ± 0.0927 0.2306 ± 0.1191
AL123456.3, GC=64.69%
Synechocystis sp. PCC 7338 0.1040 ± 0.0443 0.1178 ± 0.0523 0.2786 ± 0.1488 0.5136 ± 0.2643 0.6361 ± 0.2206
CP054306.1, GC=47.14%
Synechocystis sp. PCC 6803 0.1210 ± 0.0789 0.1012 ± 0.0544 0.2226 ± 0.1035 0.4817 ± 0.2427 0.5450 ± 0.1818
NC 000911.1, GC=47.04%
Streptococcus agalactiae NEM316 0.0188 ± 0.0062 0.2402 ± 0.1256 0.5596 ± 0.2656 0.7715 ± 0.3196 0.8209 ± 0.3002
NC 004368.1, GC=35.12%
Overall mean recall 0.15 ± 0.10 0.13 ± 0.09 0.25 ± 0.20 0.46 ± 0.23 0.57 ± 0.21
Table 9: Average recall over ten overlap thresholds
on two archaeal species of the two best perform-
ing terminator prediction tools (TermNN and Bac-
TermFinder) as per the results shown in Table 8. The
highest recall per Archaea is highlighted in bold.
Archaea
TermNN
BacTermFinder
Haloferax volcanii
0.1536 ± 0.1067
0.3158 ± 0.1969
NC
013967, GC=65.69%
Methanococcus maripaludis
0.4000 ± 0.2235
0.5712 ± 0.2468
NC
00579, GC=32.63%
ability of being a terminator outputted by TermNN
and BacTermFinder. To do this, we sorted their pre-
dictions based on their estimated probabilities of a
sequence being a terminator, and selected the top
n% to calculate recall at ten different percentage
overlaps with actual terminators. Subsequently, we
calculated the mean recall and standard deviation
across the percentage overlap levels. We visualized
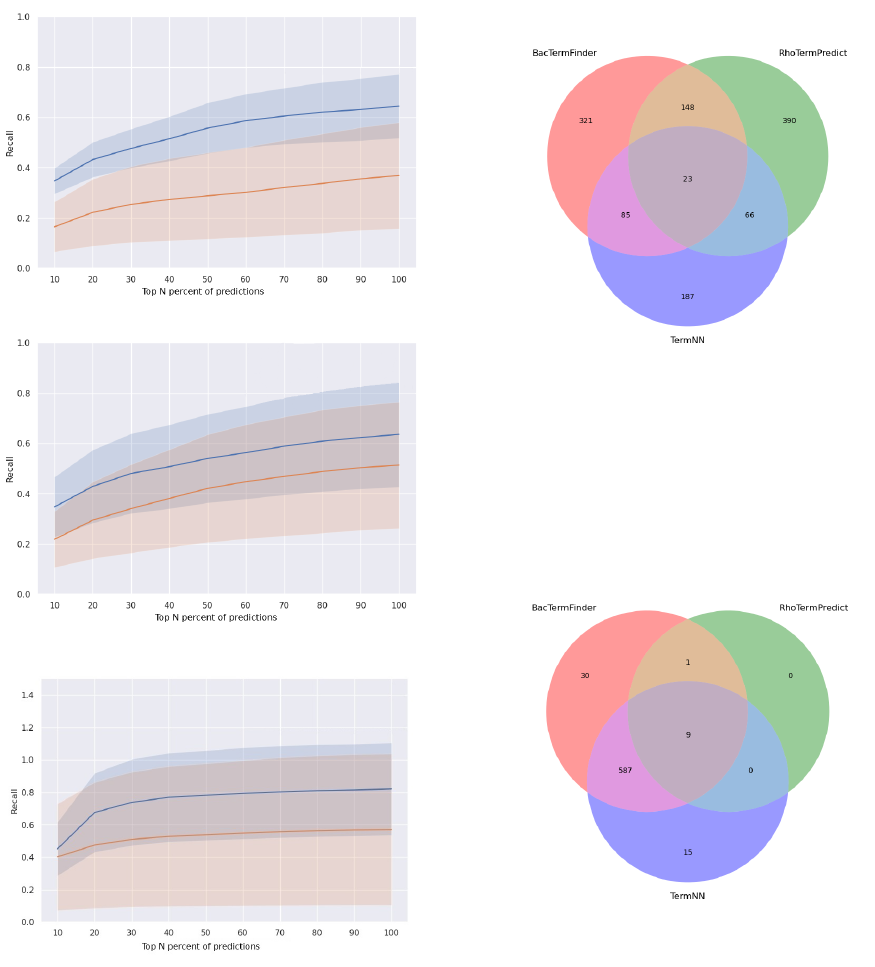
mean recall vs top n predictions for a bacterium with
high GC content (S. gardneri, Fig. 11(A)), a bac-
terium with approximately equiprobable nucleotide
distribution (Synechocystis PCC 7338, Fig. 11(B)),
and a bacterium with low GC content (S. gardneri,
Fig. 11(C)). For these three bacteria, BacTermFinder
achieves higher recall rates at any top n% predictions
than TermNN. BacTermFinder has a wider margin
over TermNN as the GC content increases. Bac-
TermFinder has also less variation in recall across
different overlap thresholds (shaded area in Fig. 11)
than TermNN. For S. gardneri, the worst recall level
of BacTermFinder is similar to or higher than the
best recall level of TermNN across all top n% of pre-
dictions. With BacTermFinder’s 10% most confident
predictions on any of these three bacteria, one is able
to identify close to 40% of their known terminators.
3.5.2 Low agreement between terminator
prediction tools
To see the agreement among RhoTermPredict,
TermNN and BacTermFinder, we generated a Venn
diagram for two bacteria in the extremes of the GC
content axis in our validation data. That is M. tu-
berculosis with 64.69% GC content and S. agalactiae
with 35.12% GC content. We chose M. tuberculosis as
it has a large proportion of factor-dependent termina-
tors [17]. For M. tuberculosis, the highest agreement
(14% of the total predicted terminators) is between
BacTermFinder and RhoTermPredict (Fig. 12, yellow
and gray areas on the center). For S. agalactiae, a low
GC bacteria, the highest agreement (92.8% of the to-
tal predicted terminators) is between BacTermFinder
and TermNN (Fig. 13, pink and gray areas on the left
side). This suggests that there is lower agreement
among tools to predict factor-dependent terminators
(Fig. 12) than intrinsic terminators (Fig. 13). Bac-
14
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint

Figure 10: Recall as a function of percentage sequence overlap between predicted and actual terminators.
All sequences are 100 nts long. The dotted lines are TermNN, and the solid lines are BacTermFinder. Each
colour represents a bacterium in our independent validation data. The area under the curves is shown in
the legend.
TermFinder predicts a similar number of terminators
in M. tuberculosis and S. agalactiae as RhoTermPre-
dict and TermNN, respectively. This suggests that
BacTermFinder is as effective in identifying factor-
dependent and intrinsic terminators as tools special-
ized on each terminator type. The number of ter-
minators predicted by all three tools is quite low for
both bacteria: 23 (or 1.9%) and 9 (or 1.4%) for M.
tuberculosis and S. agalactiae, respectively.
4 Conclusions
In this work, we have collected, to our knowledge,
the largest data set of bacterial terminators identi-
fied by sequencing technologies. This comprehensive
dataset includes terminators from 25 bacterial strains
with a wide range of GC content identified by vari-
ous sequencing technologies. We expect these data to
be valuable for further developments in bacterial ter-
minator prediction. Additionally, we have developed
BacTermFinder, a general model for finding bacterial
terminators. Future work to improve BacTermFinder
includes 1) increasing the region of interest to include
the Rho utilization site (rut); 2) adding archaeal ter-
minators into the training data; and 3) determining
the terminator type, as BacTermFinder can find both
terminator types but does not indicate which type is
found.
BacTermFinder outperforms in terms of recall
other existing tools in an independent validation
data set. BacTermFinder’s average recall over five
bacterial species is 0.57 ± 0.21, and TermNN’s
(the second-best tool) recall is 0.46 ± 0.23. This
increase in recall is achieved by BacTermFinder
while predicting, on average, two terminators less
per gene than TermNN. BacTermFinder recall in all
prokaryotic data (bacterial and archaeal) is 0.53 ±
0.20, and TermNN’s is 0.40 ± 0.22. Furthermore,
BacTermFinder can identify both types of termina-
tors as good as or better than tools specialized on
that specific terminator type. BacTermFinder and
BacTermData are available at:
https://github.com/BioinformaticsLabAtMUN/BacTermFinder.
15
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint
A
B
C
Figure 11: Average recall (solid line) versus top
n% most confident predictions for BacTermFinder
(blue) and TernNN (orange) for A S. gardneri, B
Synechocystis PCC 7338, and C S. agalactiae. The
shaded area indicates standard deviation.
Figure 12: Agreement among 1,220 predicted termi-
nators (with at least 50% sequence overlap with an
actual terminator) of BacTermFinder, RhoTermPre-
dict and TermNN for M. tuberculosis.
Figure 13: Agreement among 642 predicted termi-
nators (with at least 50% sequence overlap with an
actual terminator) of BacTermFinder, RhoTermPre-
dict and TermNN for S.agalactiae.
16
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint
5 Competing interests
The authors do not have any competing interests.
6 Author contributions state-
ment
L.P-C. conceived the project, supervised it, in-
terpreted the results, wrote and reviewed the
manuscript. S.M.A.T.G. collected BacTermData,
implemented BacTermFinder, conducted the exper-
iments, analysed and interpreted the results, wrote
and reviewed the manuscript.
7 Acknowledgments
This research was partly enabled by computing in-
frastructure provided by Acenet (ace-net.ca) and the
Digital Research Alliance of Canada (alliancecan.ca).
This work was supported by funds from an NSERC
Discovery Grant to L.P-C. and a graduate fellowship
from Memorial University School of Graduate Stud-
ies to S.M.A.T.G.
References
[1] National center for biotechnology information
(NCBI) pubmed. https://pubmed.ncbi.nlm.
nih.gov/, [1988] – [2023]. Accessed: 2023-11-
10.
[2] National center for biotechnology informa-
tion (NCBI) gene expression omnibus (GEO).
https://www.ncbi.nlm.nih.gov/geo/, 1999 –
[2023]. Accessed: 2023-11-10.
[3] Philip P. Adams, Gabriele Baniulyte, Caro-
line Esnault, Kavya Chegireddy, Navjot Singh,
Molly Monge, Ryan K. Dale, Gisela Storz, and
Joseph T. Wade. Regulatory roles of Escherichia
coli 5’ UTR and ORF-internal RNAs detected
by 3’ end mapping. eLife, 10:1–33, 1 2021.
[4] Sharmistha Banerjee, Jisha Chalissery, Irfan
Bandey, and Ranjan Sen. Rho-dependent tran-
scription termination: more questions than an-
swers. Journal of microbiology (Seoul, Korea),
44(1):11, 2006.
[5] Laur`ene Bastet, Pilar Bustos-Sanmamed, Aran-
cha Catalan-Moreno, Carlos J. Caballero, Ser-
gio Cuesta, Leticia Matilla-Cuenca, Maite Vil-
lanueva, Jaione Valle, I˜nigo Lasa, and Alejan-
dro Toledo-Arana. Regulation of heterogenous
LexA expression in Staphylococcus aureus by an
antisense RNA originating from transcriptional
read-through upon natural mispairings in the
sbrB intrinsic terminator. International Journal
of Molecular Sciences, 23, 1 2022.
[6] Sarah J. Berkemer, Lisa Katharina Maier,
Fabian Amman, Stephan H. Bernhart, Julia
W¨ortz, Pascal M¨arkle, Friedhelm Pfeiffer, Pe-
ter F. Stadler, and Anita Marchfelder. Identifi-
cation of RNA 3´ ends and termination sites in
Haloferax volcanii. RNA Biology, 17:663, 5 2020.
[7] Robson P. Bonidia, Douglas S. Domingues,
Danilo S. Sanches, and Andr´e C.P.L.F. De Car-
valho. MathFeature: feature extraction package
for DNA, RNA and protein sequences based on
mathematical descriptors. Briefings in Bioinfor-
matics, 23:1–10, 1 2022.
[8] Vivian B. Brandenburg, Franz Narberhaus, and
Axel Mosig. Inverse folding based pre-training
for the reliable identification of intrinsic tran-
scription terminators. PLOS Computational Bi-
ology, 18:e1010240, 7 2022.
[9] Zhen Chen, Pei Zhao, Chen Li, Fuyi Li, Dongxu
Xiang, Yong Zi Chen, Tatsuya Akutsu, Roger J.
Daly, Geoffrey I. Webb, Quanzhi Zhao, Lukasz
Kurgan, and Jiangning Song. iLearnPlus: a
comprehensive and automated machine-learning
platform for nucleic acid and protein sequence
analysis, prediction and visualization. Nucleic
Acids Research, 49:e60–e60, 6 2021.
[10] Ruben Chevez-Guardado and Lourdes Pe˜na-
Castillo. Promotech: a general tool for bacterial
17
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint
promoter recognition. Genome Biology, 22:1–16,
12 2021.
[11] Sang-Hyeok Cho, Yujin Jeong, Seong-Joo Hong,
Hookeun Lee, Hyung-Kyoon Choi, Dong-Myung
Kim, Choul-Gyun Lee, Suhyung Cho, and
Byung-Kwan Cho. Different regulatory modes
of Synechocystis sp. PCC 6803 in response to
photosynthesis inhibitory conditions. mSystems,
6, 12 2021.
[12] Donghui Choe, Kangsan Kim, Minjeong Kang,
Seung Goo Lee, Suhyung Cho, Bernhard Pals-
son, and Byung Kwan Cho. Synthetic 3’-UTR
valves for optimal metabolic flux control in Es-
cherichia coli. Nucleic Acids Research, 50:4171,
4 2022.
[13] Daniel Dar, Daniela Prasse, Ruth A. Schmitz,
and Rotem Sorek. Widespread formation of al-
ternative 3’ UTR isoforms via transcription ter-
mination in archaea. Nature Microbiology 2016
1:10, 1:1–9, 8 2016.
[14] Daniel Dar, Maya Shamir, J. R. Mellin, Mikael
Koutero, Noam Stern-Ginossar, Pascale Cossart,
and Rotem Sorek. Term-seq reveals abundant
ribo-regulation of antibiotics resistance in bac-
teria. Science, 352(6282):aad9822, 2016.
[15] Daniel Dar and Rotem Sorek. High-resolution
RNA 3’-ends mapping of bacterial Rho-
dependent transcripts. Nucleic Acids Research,
46:6797, 7 2018.
[16] Marco Di Salvo, Simone Puccio, Clelia
Peano, Stephan Lacour, and Pietro Alifano.
RhoTermPredict: an algorithm for predict-
ing Rho-dependent transcription terminators
based on Escherichia coli, Bacillus subtilis and
Salmonella enterica databases. BMC bioinfor-
matics, 20(1):117, March 2019.
[17] Alexandre D’Halluin, Peter Polgar, Terry Kipko-
rir, Zaynah Patel, Teresa Cortes, and Kristine B.
Arnvig. Premature termination of transcription
is shaped by Rho and translated uORFS in My-
cobacterium tuberculosis. iScience, 26(4):106465,
2023.
[18] Yongxian Fan, Wanru Wang, and Qingqi Zhu.
iterb-PPse: Identification of transcriptional
terminators in bacterial by incorporating nu-
cleotide properties into PseKNC. PloS One,
15(5):e0228479, 2020.
[19] Chao-Qin Feng, Zhao-Yue Zhang, Xiao-Juan
Zhu, Yan Lin, Wei Chen, Hua Tang, and Hao
Lin. iTerm-PseKNC: a sequence-based tool for
predicting bacterial transcriptional terminators.
Bioinformatics, 35(9):1469–1477, May 2019.
[20] Rapha¨el Forquet, Xuejiao Jiang, William
Nasser, Florence Hommais, Sylvie Reverchon,
and Sam Meyer. Mapping the complex tran-
scriptional landscape of the Phytopathogenic
bacterium Dickeya dadantii. mBio, 13, 6 2022.
[21] Manuela Fuchs, Vanessa Lamm-Schmidt, Jo-
hannes Sulzer, Falk Ponath, Laura Jenniches,
Joseph A. Kirk, Robert P. Fagan, Lars Barquist,
J¨org Vogel, and Franziska Faber. An RNA-
centric global view of Clostridioides difficile re-
veals broad activity of Hfq in a clinically im-
portant gram-positive bacterium. Proceedings of
the National Academy of Sciences of the United
States of America, 118, 6 2021.
[22] Socorro Gama-Castro, Heladia Salgado, Alberto
Santos-Zavaleta, Daniela Ledezma-Tejeida, Luis
Mu˜niz-Rascado, Jair Santiago Garc´ıa-Sotelo,
Kevin Alquicira-Hern´andez, Irma Mart´ınez-
Flores, Lucia Pannier, Jaime Abraham Castro-
Mondrag´on, Alejandra Medina-Rivera, Hilda
Solano-Lira, C´esar Bonavides-Mart´ınez, Ernesto
P´erez-Rueda, Shirley Alquicira-Hern´andez, Lil-
iana Porr´on-Sotelo, Alejandra L´opez-Fuentes,
Anastasia Hern´andez-Koutoucheva, V´ıctor Del
Moral-Chavez, Fabio Rinaldi, and Julio Collado-
Vides. RegulonDB version 9.0: high-level inte-
gration of gene regulation, coexpression, motif
clustering and beyond. Nucleic Acids Research,
44:D133–D143, 1 2016.
[23] Paul P. Gardner, Lars Barquist, Alex Bateman,
Eric P. Nawrocki, and Zasha Weinberg. RNIE:
18
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint
genome-wide prediction of bacterial intrinsic ter-
minators. Nucleic Acids Research, 39(14):5845–
5852, August 2011.
[24] Adrian Sven Geissler, Christian Anthon, Ferhat
Alkan, Enrique Gonz´alez-Tortuero, Line Dahl
Poulsen, Thomas Beuchert Kallehauge, Anne
Bre¨uner, Stefan Ernst Seemann, Jeppe Vinther,
and Jan Gorodkin. BSGatlas: a unified Bacil-
lus subtilis genome and transcriptome annota-
tion atlas with enhanced information access. Mi-
crobial Genomics, 7:524, 2021.
[25] Leo Grinsztajn, Edouard Oyallon, and Gael
Varoquaux. Why do tree-based models still out-
perform deep learning on typical tabular data?,
6 2022.
[26] Swati Gupta and Debnath Pal. Clusters of hair-
pins induce intrinsic transcription termination in
bacteria. Scientific Reports, 11(1):16194, August
2021.
[27] Michael J.L. De Hoon, Yuko Makita, Kersta
Nakai, and Satora Msyasio. Prediction of tran-
scriptional terminators in Bacillus subtilis and
related species. PLoS Computational Biology,
1:0212–0221, 2 2005.
[28] Soonkyu Hwang, Namil Lee, Donghui Choe,
Yongjae Lee, Woori Kim, Yujin Jeong, Suhyung
Cho, Bernhard O. Palsson, and Byung-
Kwan Cho. Elucidating the regulatory ele-
ments for transcription termination and post-
transcriptional processing in the Streptomyces
clavuligerus genome. mSystems, 6, 6 2021.
[29] Soonkyu Hwang, Namil Lee, Donghui Choe,
Yongjae Lee, Woori Kim, Ji Hun Kim, Gahyeon
Kim, Hyeseong Kim, Neung Ho Ahn, By-
oung Hee Lee, Bernhard O. Palsson, and
Byung Kwan Cho. System-level analysis of tran-
scriptional and translational regulatory elements
in Streptomyces griseus. Frontiers in Bioengi-
neering and Biotechnology, 10, 2 2022.
[30] Takahiro Ishii, Ken Ichi Yoshida, Goro Terai,
Yasutaro Fujita, and Kenta Nakai. DBTBS:
a database of Bacillus subtilis promoters and
transcription factors. Nucleic Acids Research,
29:278–280, 1 2001.
[31] Yujin Jeong, Seong Joo Hong, Sang Hyeok Cho,
Seonghoon Yoon, Hookeun Lee, Hyung Ky-
oon Choi, Dong Myung Kim, Choul Gyun
Lee, Suhyung Cho, and Byung Kwan Cho.
Multi-omic analyses reveal habitat adaptation of
marine cyanobacterium Synechocystis sp. PCC
7338. Frontiers in Microbiology, 12, 5 2021.
[32] Grace E. Johnson, Jean Benoˆıt Lalanne,
Michelle L. Peters, and Gene Wei Li. Func-
tionally uncoupled transcription-translation in
Bacillus subtilis. Nature, 585:124, 9 2020.
[33] Xiangwu Ju, Dayi Li, and Shixin Liu. Full-
length RNA profiling reveals pervasive bidirec-
tional transcription terminators in bacteria. Na-
ture microbiology, 4:1907, 11 2019.
[34] Mohamad Al kadi, Eiji Ishii, Dang Tat Truong,
Daisuke Motooka, Shigeaki Matsuda, Tetsuya
Iida, Toshio Kodama, and Daisuke Okuzaki. Di-
rect RNA sequencing unfolds the complex tran-
scriptome of Vibrio parahaemolyticus. mSys-
tems, 6, 12 2021.
[35] Guolin Ke, Qi Meng, Thomas Finley, Taifeng
Wang, Wei Chen, Weidong Ma, Qiwei Ye, and
Tie-Yan Liu. LightGBM: A highly efficient gra-
dient boosting decision tree. In I. Guyon, U. Von
Luxburg, S. Bengio, H. Wallach, R. Fergus,
S. Vishwanathan, and R. Garnett, editors, Ad-
vances in Neural Information Processing Sys-
tems, volume 30. Curran Associates, Inc., 2017.
[36] Carleton L. Kingsford, Kunmi Ayanbule, and
Steven L. Salzberg. Rapid, accurate, computa-
tional discovery of Rho-independent transcrip-
tion terminators illuminates their relationship to
DNA uptake. Genome Biology, 8:1–12, 2 2007.
[37] Thomas Kluyver, Benjamin Ragan-Kelley, Fer-
nando P´erez, Brian Granger, Matthias Busson-
nier, Jonathan Frederic, Kyle Kelley, Jessica
Hamrick, Jason Grout, Sylvain Corlay, Paul
19
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint
Ivanov, Dami´an Avila, Safia Abdalla, and Carol
Willing. Jupyter notebooks – a publishing for-
mat for reproducible computational workflows.
In F. Loizides and B. Schmidt, editors, Position-
ing and Power in Academic Publishing: Players,
Agents and Agendas, pages 87 – 90. IOS Press,
2016.
[38] Jean Benoˆıt Lalanne, James C. Taggart, Mon-
ica S. Guo, Lydia Herzel, Ariel Schieler, and
Gene Wei Li. Evolutionary convergence of
pathway-specific enzyme expression stoichiome-
try. Cell, 173:749–761.e38, 4 2018.
[39] Jean Benoˆıt Lalanne, James C. Taggart, Mon-
ica S. Guo, Lydia Herzel, Ariel Schieler, and
Gene Wei Li. Evolutionary convergence of
pathway-specific enzyme expression stoichiome-
try. Cell, 173:749, 4 2018.
[40] Yann LeCun, Y. Bengio, and Geoffrey Hinton.
Deep learning. Nature, 521:436–44, 05 2015.
[41] Yongjae Lee, Namil Lee, Soonkyu Hwang, Woori
Kim, Suhyung Cho, Bernhard O. Palsson, and
Byung Kwan Cho. Genome-scale analysis of ge-
netic regulatory elements in Streptomyces aver-
mitilis MA-4680 using transcript boundary in-
formation. BMC Genomics, 23, 12 2022.
[42] Yongjae Lee, Namil Lee, Soonkyu Hwang, Woori
Kim, Yujin Jeong, Suhyung Cho, Bernhard O.
Palsson, and Byung Kwan Cho. Genome-scale
determination of 5´ and 3´ boundaries of RNA
transcripts in Streptomyces genomes. Scientific
Data, 7, 12 2020.
[43] Yongjae Lee, Namil Lee, Yujin Jeong, Soonkyu
Hwang, Woori Kim, Suhyung Cho, Bernhard O.
Palsson, and Byung Kwan Cho. The transcrip-
tion unit architecture of Streptomyces lividans
TK24. Frontiers in Microbiology, 10:2074, 9
2019.
[44] Elena A. Lesnik, Rangarajan Sampath,
Harold B. Levene, Timothy J. Henderson,
John A. McNeil, and David J. Ecker. Prediction
of rho-independent transcriptional terminators
in Escherichia coli. Nucleic Acids Research,
29(17):3583–3594, 09 2001.
[45] Jie Li, Lei Yue, Zhihua Li, Wenting Zhang,
Bing Zhang, Fangqing Zhao, and Xiuzhu Dong.
aCPSF1 cooperates with terminator U-tract to
dictate archaeal transcription termination effi-
cacy. eLife, 10:70464, 12 2021.
[46] Bin Liu, Fule Liu, Longyun Fang, Xiaolong
Wang, and Kuo Chen Chou. repDNA: a
Python package to generate various modes of
feature vectors for DNA sequences by incorpo-
rating user-defined physicochemical properties
and sequence-order effects. Bioinformatics (Ox-
ford, England), 31:1307–1309, 4 2015.
[47] Scott M. Lundberg, Gabriel Erion, Hugh
Chen, Alex DeGrave, Jordan M. Prutkin, Bala
Nair, Ronit Katz, Jonathan Himmelfarb, Nisha
Bansal, and Su-In Lee. From local explanations
to global understanding with explainable ai for
trees. Nature Machine Intelligence, 2(1):2522–
5839, 2020.
[48] Antonio Majdandzic, Chandana Rajesh, and Pe-
ter K. Koo. Correcting gradient-based interpre-
tations of deep neural networks for genomics.
Genome Biology, 24(1):109, 2023.
[49] Zachary F. Mandell, Reid T. Oshiro, Alexan-
der V. Yakhnin, Rishi Vishwakarma, Mikhail
Kashlev, Daniel B. Kearns, and Paul Babitzke.
NusG is an intrinsic transcription termination
factor that stimulates motility and coordinates
gene expression with NusA. eLife, 10, 2021.
[50] Daniel G. Mediati, Julia L. Wong, Wei Gao, Stu-
art McKellar, Chi Nam Ignatius Pang, Sylva-
nia Wu, Winton Wu, Brandon Sy, Ian R. Monk,
Joanna M. Biazik, Marc R. Wilkins, Benjamin P.
Howden, Timothy P. Stinear, Sander Granne-
man, and Jai J. Tree. RNase III-CLASH of
multi-drug resistant Staphylococcus aureus re-
veals a regulatory mRNA 3’UTR required for in-
termediate vancomycin resistance. Nature Com-
munications, 13, 12 2022.
20
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint
[51] Adi Millman, Daniel Dar, Maya Shamir, and
Rotem Sorek. Computational prediction of reg-
ulatory, premature transcription termination in
bacteria. Nucleic Acids Research, 45(2):886–893,
January 2017.
[52] C´edric Nadiras, Eric Eveno, Annie Schwartz,
Nara Figueroa-Bossi, and Marc Boudvillain.
A multivariate prediction model for Rho-
dependent termination of transcription. Nu-
cleic Acids Research, 46(16):8245–8260, Septem-
ber 2018.
[53] The pandas development team. pandas-
dev/pandas: Pandas, February 2020. https:
//doi.org/10.5281/zenodo.3509134.
[54] Aaron R. Quinlan and Ira M. Hall. BEDTools:
a flexible suite of utilities for comparing ge-
nomic features. Bioinformatics (Oxford, Eng-
land), 26:841–842, 1 2010.
[55] Ananya Ray-Soni, Michael J. Bellecourt, and
Robert Landick. Mechanisms of bacterial tran-
scription termination: All good things must
end. https://doi.org/10.1146/annurev-biochem-
060815-014844, 85:319–347, 6 2016.
[56] Isabelle Rosinski-Chupin, Elisabeth Sauvage,
Odile Sismeiro, Adrien Villain, Violette Da
Cunha, Marie Elise Caliot, Marie Agn`es Dil-
lies, Patrick Trieu-Cuot, Philippe Bouloc,
Marie Fr´ed´erique Lartigue, and Philippe Glaser.
Single nucleotide resolution RNA-seq uncovers
new regulatory mechanisms in the opportunis-
tic pathogen Streptococcus agalactiae. BMC Ge-
nomics, 16, 5 2015.
[57] Thomas J. Santangelo and Irina Artsimovitch.
Termination and antitermination: RNA poly-
merase runs a stop sign. Nature Reviews Mi-
crobiology 2011 9:5, 9:319–329, 4 2011.
[58] Alberto Santos-Zavaleta, Heladia Salgado,
Socorro Gama-Castro, Mishael S´anchez-P´erez,
Laura G´omez-Romero, Daniela Ledezma-
Tejeida, Jair Santiago Garc´ıa-Sotelo, Kevin
Alquicira-Hern´andez, Luis Jos´e Mu˜niz-Rascado,
Pablo Pe˜na-Loredo, Cecilia Ishida-Guti´errez,
David A. Vel´azquez-Ram´ırez, V´ıctor Del
Moral-Ch´avez, C´esar Bonavides-Mart´ınez,
Carlos Francisco M´endez-Cruz, James Galagan,
and Julio Collado-Vides. RegulonDB v 10.5:
tackling challenges to unify classic and high
throughput knowledge of gene regulation in E.
coli K-12. Nucleic Acids Research, 47:D212, 1
2019.
[59] Cynthia M. Sharma and J¨org Vogel. Differential
RNA-seq: the approach behind and the biologi-
cal insight gained. Current Opinion in Microbi-
ology, 19:97–105, 6 2014.
[60] Alexander A. Shishkin, Georgia Giannoukos,
Alper Kucukural, Dawn Ciulla, Michele Busby,
Christine Surka, Jenny Chen, Roby P. Bhat-
tacharyya, Robert F. Rudy, Milesh M. Patel,
Nathaniel Novod, Deborah T. Hung, Andreas
Gnirke, Manuel Garber, Mitchell Guttman, and
Jonathan Livny. Simultaneous generation of
many RNA-seq libraries in a single reaction. Na-
ture methods, 12:323, 3 2015.
[61] Jelle Slager, Rieza Aprianto, and Jan Willem
Veening. Deep genome annotation of the oppor-
tunistic human pathogen Streptococcus pneumo-
niae D39. Nucleic Acids Research, 46:9971, 11
2018.
[62] Eunho Song, Heesoo Uhm, Palinda Ruvan
Munasingha, Seungha Hwang, Yeon-Soo Seo,
Jin Young Kang, Changwon Kang, and Sungchul
Hohng. Rho-dependent transcription termina-
tion proceeds via three routes. Nature Commu-
nications, 13(1):1663, 2022.
[63] Hiraku Takada, Zachary F. Mandell, Helen
Yakhnin, Anastasiya Glazyrina, Shinobu Chiba,
Tatsuaki Kurata, Kelvin J.Y. Wu, Ben I.C.
Tresco, Andrew G. Myers, Gemma C. Aktinson,
Paul Babitzke, and Vasili Hauryliuk. Expression
of Bacillus subtilis ABCF antibiotic resistance
factor vmlr is regulated by RNA polymerase
pausing, transcription attenuation, translation
attenuation and (p)ppgpp. Nucleic Acids Re-
search, 50:6174, 6 2022.
21
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint
[64] Maureen K. Thomason, Maya Voichek, Daniel
Dar, Victoria Addis, David Fitzgerald, Susan
Gottesman, Rotem Sorek, and E. Peter Green-
berga. A rhli 5’ UTR-derived sRNA regu-
lates RhlR-dependent quorum sensing in Pseu-
domonas aeruginosa. mBio, 10, 2019.
[65] Jessica M. Vera, Indro Neil Ghosh, Yaoping
Zhang, Alex S. Hebert, Joshua J. Coon, and
Robert Landick. Genome-scale transcription-
translation mapping reveals features of Zy-
momonas mobilis transcription units and pro-
moters. mSystems, 5, 8 2020.
[66] Indu Warrier, Nikhil Ram-Mohan, Zeyu Zhu,
Ariana Hazery, Haley Echlin, Jason Rosch,
Michelle M. Meyer, and Tim van Opijnen. The
transcriptional landscape of Streptococcus pneu-
moniae TIGR4 reveals a complex operon archi-
tecture and abundant riboregulation critical for
growth and virulence. PLoS Pathogens, 14, 12
2018.
[67] E. V. Wong and Open Textbook Library. Cells :
molecules and mechanisms. pages 1–276, 2009.
[68] Qing Song Xu and Yi Zeng Liang. Monte carlo
cross validation. Chemometrics and Intelligent
Laboratory Systems, 56:1–11, 4 2001.
[69] Bo Yan, Matthew Boitano, Tyson A. Clark, and
Laurence Ettwiller. SMRT-Cappable-seq reveals
complex operon variants in bacteria. Nature
Communications, 9, 12 2018.
22
.CC-BY-NC 4.0 International licenseavailable under a
was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprint (whichthis version posted July 8, 2024. ; https://doi.org/10.1101/2024.07.05.602086doi: bioRxiv preprint
